MULTISYSTÉMOVÁ ATROFIE
Multisystémová atrofie (MSA) je progresivní neurodegenerativní onemocnění se sporadickým výskytem, patřící do skupiny synukleinopatií (kam řadíme i Parkinsonovu nemoc a demenci s Lewyho tělísky). Klinické projevy zahrnují mozečkové, parkinsonské, pyramidové i sfinkterové obtíže a dysautonomii, v různých kombinacích a v různé intenzitě.
Průběh je zpravidla pozvolna progredující, onemocnění začíná převážně ve věku 45-65 let, muži i ženy bývají postižení stejně často.
Parkinsonské projevy u pacientů s MSA probíhají většinou pod obrazem bradykineze, rigidity, posturální instability a dysartrie. Třes může být přítomen, nemívá však ráz klidového tremoru typického pro Parkinsonovu nemoc, spíše bývá posturální nebo intenční, často asymetrický a někdy i s myokloniemi. Odpověď na léčbu levodopou je zpravidla velmi omezená.
Hlavním mozečkovým postižením u MSA jsou poruchy chůze a stability, vzácností nejsou hypermetrie, dysmetrie, intenční tremor a poruchy okulomotoriky s hypermetrickými sakádami, někteří pacienti mohou mít mozečkovou dysartrii se skandovanou řečí.
Pyramidová dysfunkce se většinou omezuje na hyperreflexii a Babinského příznak.
Typická pro MSA je dysautonomie a výskyt sfinkterových obtíží. Ortostatická hypotenze může být zjevná, vedoucí až ke kolapsovým stavům, v některých případech ale může být i asymptomatická. Klinicky ji lze charakterizovat poklesem systolického tlaku o 30 torr nebo diastolického o 15 torr do tří minut po prudké vertikalizaci z lehu. Častým doprovodným znakem je nedostatečná kompenzatorní tachykardie (při vertikalizaci chybějící tlaková adaptace není kompenzována odpovídajícím urychlením pulsu - tepová frekvence naroste o méně než 10 tepů/min).
Inkontinence, imperativní mikce a poruchy potence mohou dotvářet klinický obraz MSA.
Mnohdy lze prokázat postižení frontálních exekutivních funkcí, většinou mírného stupně. Naproti tomu plně vyvinutá demence nepatří do typického obrazu MSA a její přítomnost by měla zaměřit diagnostické pátrání jiným směrem.
Z pomocných metod může být
užitečná MRI s relativně častým nálezem atrofie mozečku a bracha pontis,
někdy lze najít i snížení signálu putamen v T2 vážených sekvencích.
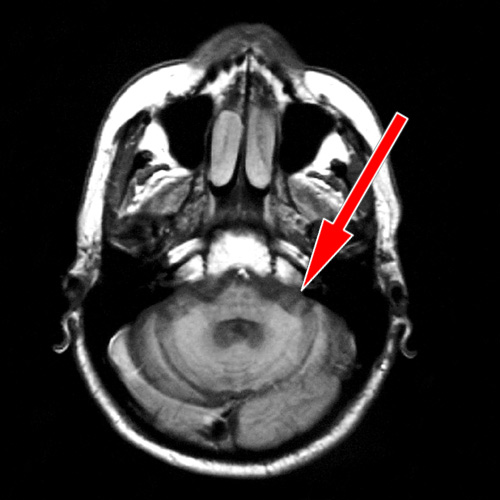
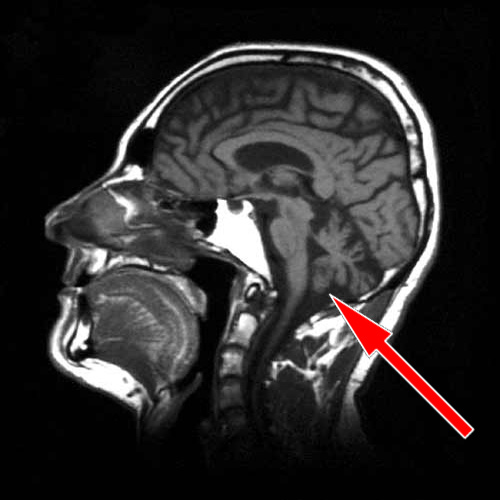
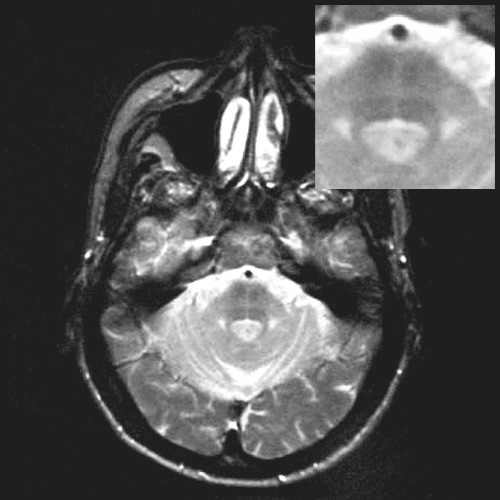
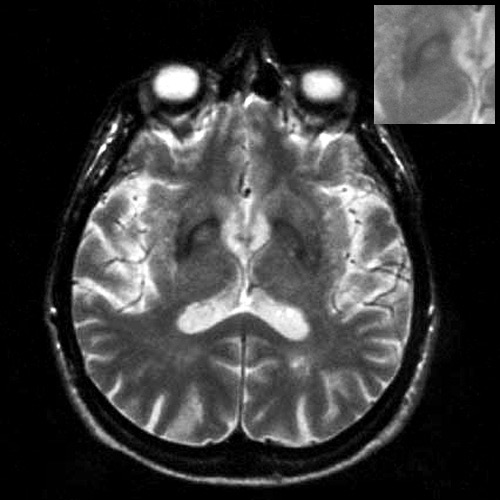
Přínosný
je EMG průkaz denervací v m. sphincter ani externus a vhodné je i vyšetření
autonomních funkcí.
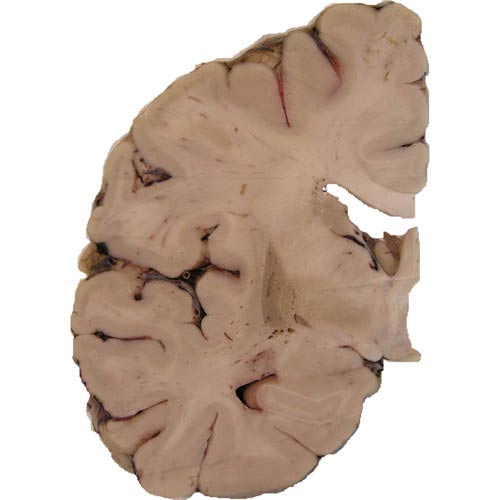
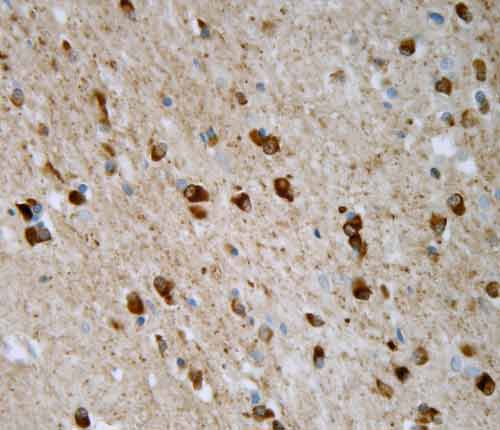
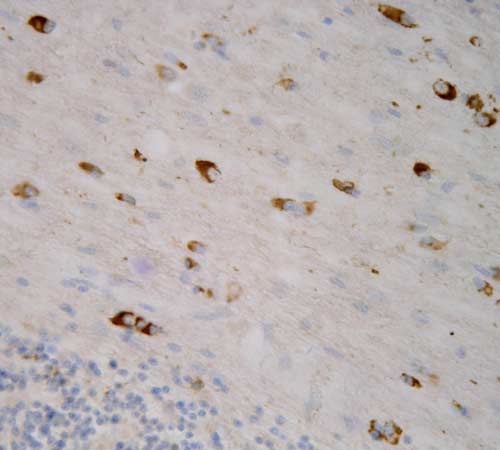
Neuropatologickým vyšetřením mozku lze již makroskopicky pozorovat nápadnou atrofii bazálních ganglií s typickým šedozeleným zbarvením putamen. V mikroskopickém obraze dominuje numerická neuronální atrofie a reaktivní glióza v mozečku, bazálních gangliích a pontu. Základním neuropatologickým diagnostickým znakem je přítomnost specifických inkluzí v neuronálních elementech a gliích. Inkluze je možné ozřejmit impregnací solemi stříbra a imunohistochemickou reakcí s protilátkami proti alfa-synukleinu a ubiquitinu.
 |
 |
 |
Dřívější členění na striatonigrální degeneraci (SND), sporadickou olivopontocerebellární atrofii (OPCA) a Shy-Dragerův syndrom se již nepoužívá, v současné době rozlišujeme pouze formu parkinsonskou s převažujícím extrapyramidovým postižením (MSA-P) a formu mozečkovou (MSA-C).
Terapeutické ovlivnění by
vždy mělo zahrnovat podávání levodopy v dostatečné dávce, přínosem mohou
být někdy i dopaminergních antagonisté, důležitá je podpůrná léčba ortostatické
hypotenze (režimová opatření, fludrokortison, midodrin) rehabilitace a
případně i logopedická péče.